دیستروفی میوتونی (MD) جزئی از یک گروه از اختلالات ارثی به نام دیستروفی های عضلانی است. این اختلال شایع ترین نوع دیستروفی عضلانی است که در بزرگسالی آغاز می شود.
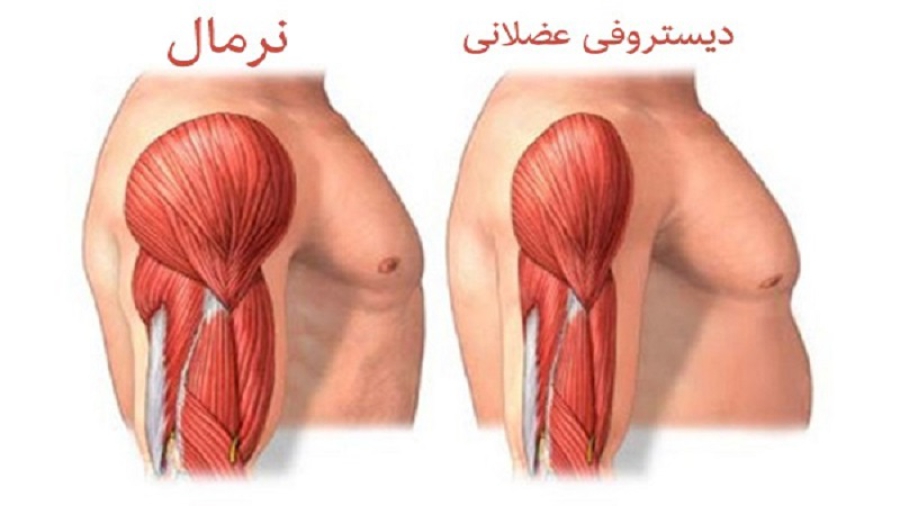
دیستروفی میوتونی با تحلیل پیشرونده و ضعف عضلات مشخص می شود. در اغلب این افراد، عضلات دچار انقباضات طولانی (میوتونی-myotonia) شده و همچنین در بعضی از عضلات امکان برگشت به حالت استراحت پس از انقباض وجود ندارد. برای مثال ممکن است افراد مبتلا در رها کردن دستگیرهِ در دچار مشکل شوند. همچنین ممکن است در این افراد لکنت زبان و یا قفل شدن موقت فک نیز مشاهده شود.
از سایر علائم MD می توان به کدورت عدسی چشم ها (آب مروارید-cataracts) و نقایص هدایتی قلب (cardiac conduction defects) اشاره کرد. علاوه بر این، ممکن است تغییرات هورمونی در مردان مبتلا منجر به طاسی زودهنگام و ناباروری (infertility) شود. اگرچه امکان بروز این علائم در هر سنی وجود دارد، اما معمولا طی بیست و یا سی سالگی ایجاد می شوند. شدت علائم این اختلال حتی در اعضای مبتلای یک خانواده نیز بسیار متفاوت است.
دو تیپ اصلی MD وجود دارد: تیپ یک (type 1) و تیپ دو (type 2). علائم ایجاد شده در این دو تیپ هم پوشانی دارند، اما تیپ دو خفیف تر از تیپ یک است. در تیپ یک، ضعف عضلانی به طور ویژه ای قسمت های تحتانی پا (lower legs)، دست ها، گردن و صورت را تحت تاثیر قرار می دهد اما در تیپ دو در ابتدا عضلات گردن، شانه، آرنج و ران ها تحت تاثیر قرار می گیرند.
تیپ های متفاوت MD، در اثر جهش در ژن های متفاوتی ایجاد می شوند.
نوعی از MD تیپ یک به نام MD مادرزادی (congenital myotonic dystrophy)، از بدو تولد مشخص می شود که از علائم آن می توان کاهش تونوس ماهیچه (هیپوتونی-hypotonia)، خمیدگی پاها به سمت داخل و بالا (پا چنبری-clubfoot)، اختلالات تنفسی، تاخیر در رشد و ناتوانی ذهنی را نام برد که بعضی از این مشکلات می توانند بسیار جدی و کشنده باشند.
شیوع بیماری دیستروفی
در سراسر جهان از هر 8.000 ، حداقل 1 نفر به MD مبتلا می باشد. میزان شیوع این دو تیپ MD در اقوام و نواحی جغرافیایی مختلف، متفاوت است. به نظر می رسد در بیشتر جمعیت ها، تیپ یک شایع تر از تیپ دو است.

ژنتیک بیماری دیستروفی
دیستروفی میوتونی تیپ یک در اثر گسترش تکرارهای نوکلئوتیدی CTG در ناحیه 3ʹ UTR (پایین دست ژن) DMPK بروز میکند. در افراد سالم این 3 نوکلوتید در حدود 5 تا 30 بار تکرار میشوند
اما در افراد بیمار این تکرار زیاد میشود و باعث بی ثباتی و بزرگ شدن بیش از حد این ناحیه روی کروموزم میشود. نوع بیماری به تعداد این تکرارها بستگی دارد. در افرادی که نوع مادرزادی مبتلا هستند تعداد تکرارها به بیش از 2000 کپی میرسد. در مبتلایان به فرم کلاسیک تعداد تکرارها حدوداً 100 تا 1000 کپی است.
در فرم خفیف بیماری حدود 50 تا 150 کپی وجود دارد. تعداد 35 تا 49 کپی از این تری نوکلئوتید هم به عنوان پیش موتاسیون در نظر گرفته میشوند.
دیستروفی میوتونی تیپ دو در اثر گسترش تکرارهای CCTG در داخل اینترون 1 ژن CNBP با نام دیگر ZNF9 ایجاد می شوند، با این حال عملکرد دقیق این ژن ها نامشخص است.
ممکن است پروتئین تولید شده توسط ژن DMPK در ارتباطات درون سلولی نقش داشته باشد. به نظر می رسد این پروتئین ها نقش مهمی در عملکرد صحیح سلول ها در قلب، مغز و ماهیچه های اسکلتی (که برای حرکت ازآن ها استفاده می شود) ایفا می کنند.
پروتئین تولید شده توسط ژن CNBP در درجه اول در قلب و ماهیچه های اسکلتی یافت شده و احتمالا نقش آن تنظیم عملکرد سایر ژن ها می باشد.
تغییرات مشابه در ساختار ژن های DMPK و CNBP باعث ایجاد انواع MD می شود. در هر مورد قطعه ای از DNA به طور غیر طبیعی به تعداد بسیار زیادی تکرار شده و باعث ایجاد ناحیه ناپایداری در ژن می شود.
ژن جهش یافته، نسخه طویل شده ای از mRNA را ایجاد می کند. mRNA الگوی مولکولی ژن است که به طور طبیعی باعث هدایت و راهنمایی تولید پروتئین می شود.
شکل غیر طبیعی و طویل mRNA باعث تشکیل توده هایی داخل سلول می شود که این توده ها در تولید بسیاری از پروتئین ها اختلال ایجاد می کنند. این اختلالات مانع از فعالیت و عملکرد طبیعی سلول های ماهیچه ای و سایر سلول ها و در بنابراین منجر به ایجاد علائم دیستروفی میوتونی می شوند.
به نظر میرسد در هر دو بیماری گسترش تکرار CTG (تکرار CUG بر روی mRNA) سبب ضبط پروتئینهای اتصالی به CUG (مانند پروتئین MBNL1 و CUGBP1) میشود و با ضبط این پروتئینها بر روی mRNA طویل شده، پیرایش mRNAهای دیگر دچار مشکل شده و علائم بیماری بروز میکند.
لازم به ذکر است پروتئین های اتصالی به توالی CUG مولکول mRNA در تنظیم پیرایش و پیرایش متناوب mRNA نقش دارند
توراث بیماری
هر دو تیپ MD دارای الگوی وراثتی اتوزومال غالب هستند. در این الگوی وراثتی، یک نسخه از ژن تغییر یافته در هر سلول برای ایجاد اختلال کافی است. در بیشتر موارد فرد مبتلا، دارای یک والد مبتلا است.
با انتقال MD از یک نسل به نسل دیگر، علائم آن زودتر و با شدت بیشتری ایجاد می شود. این پدیده که پیش اندازی یا انتظار (anticipation) نام دارد، در هر دو تیپ MD گزارش شده است اما به نظر می رسد این پدیده در دیستروفی میوتونی تیپ یک، قوی تر است.
علت ایجاد پیشاندازی در این اختلال، افزایش طول ناحیه ناپایدار در ژن DMPK است. امکان ایجاد پیش اندازی در MD تیپ دو و مکانیسم ایجاد آن نامشخص است. به نظر نمی رسد افزایش طول ناحیه ناپایدار در ژن CNBP، در سن ایجاد علائم زودهنگام این اختلال تاثیر گذار باشد.
نام های دیگر بیماری
dystrophia myotonica
yotonia atrophica
myotonia dystrophica


{kind=link}